




The information in this section will help you to understand the fundamentals of recognising, referring and supporting a patient with an interstitial lung disease in primary care.
This is not a substitute for completing an appropriate respiratory assessment module. For advice and support on choosing the right course for you, please see our training and development page.
Interstitial Lung Diseases
Interstitial Lung Diseases (ILDs) are a group of over 200 diseases that cause scarring (fibrosis) or inflammation in the lung parenchyma.
The lung parenchyma refers to the functional tissue of the lungs involved in gas exchange. It primarily includes:
Alveoli: The tiny air sacs where oxygen is absorbed into the bloodstream and carbon dioxide is removed.
Bronchioles: The small airways that lead to the alveoli, responsible for conducting air to the alveolar spaces.
Interstitial Tissue: The supportive connective tissue surrounding the alveoli and small airways, which provides structure to the lungs and plays a role in gas exchange.
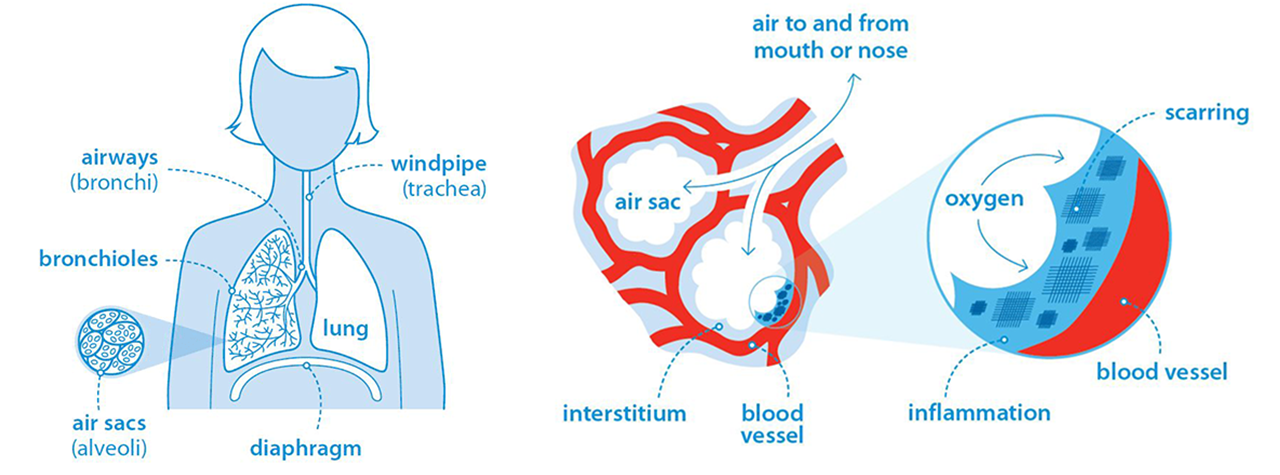
Interstitial lung diseases cause the interstitial lung tissue to become stiff and thickened, or 'scarred'. This is called pulmonary fibrosis.
The lungs lose elasticity, making it more difficult to expand fully during inspiration. This restricts deep breaths and can lead to a decrease in oxygen exchange, causing a drop in the patient's oxygen levels.
ILDs are restrictive lung conditions, because the lung volume and the ability to fully expand the lungs during inspiration is decreased. The large airways are unaffected.
They differ from obstructive lung obstructive diseases such as asthma and COPD, in which narrowed airways cause difficulty exhaling all the air from the lungs.
It is possible to have mixed obstruction and restriction if a patient has an ILD in addition to a condition such as COPD or asthma.
Some ILDs are progressive, meaning that fibrosis develops and worsens over time and some are stable. Progressive ILDs have a much poorer outlook for patients
Not all ILDs result in permanent scarring, especially if diagnosed early and treated appropriately. This is why it is important that patients in primary care are identified quickly and referred for further testing and diagnosis.
Symptoms of ILD to look out for in patients
Symptoms include:
• gradual breathlessness which is worse on exertion
• persistent cough, often dry
• fatigue
• bilateral inspiratory 'velcro' crackles when listening to the chest
• clubbing of the fingers
Causes of ILD
Only around one in three cases of interstitial lung disease has an identifiable cause. Experts suggest that a combination of genetic predisposition and environmental factors may contribute to the development of some conditions, such as sarcoidosis.
Patients who are more likely to have interstitial lung disease (ILD) tend to fall into several high-risk categories based on a combination of age, medical history, environmental exposure, and lifestyle factors. The following groups are at increased risk:
1. Older Adults (Typically Over 50 Years Old):
Idiopathic Pulmonary Fibrosis (IPF), one of the most common and severe forms of ILD, primarily affects people over 50. Age is a significant risk factor for many forms of ILD, especially IPF
2. Patients with Connective Tissue Diseases:
People with autoimmune conditions, such as rheumatoid arthritis, systemic sclerosis and systemic lupus erythematosus, are at higher risk of developing connective tissue disease-associated ILD
3. Smokers or Ex-Smokers:
Smoking significantly increases the risk of developing certain types of ILD, including respiratory bronchiolitis-associated ILD and idiopathic pulmonary fibrosis.
4. Occupational and Environmental Exposures:
Patients with long-term exposure to certain inhalants, such as asbestos, silica, coal dust, mold, and bird proteins, are at risk of developing forms of ILD like asbestosis, hypersensitivity pneumonitis, or pneumoconiosis
5. Medication-Induced ILD:
Certain medications, particularly chemotherapy drugs (e.g., bleomycin), antiarrhythmic drugs (e.g., amiodarone), and antibiotics (e.g., nitrofurantoin), are known to cause drug-induced ILD.
6. Patients with Family History of ILD:
Individuals with a family history of ILD, particularly idiopathic pulmonary fibrosis (IPF), are at greater risk of developing the disease, suggesting a potential genetic predisposition
7. Individuals with Gastroesophageal Reflux Disease (GORD):
There is a recognized association between GORD and ILD, particularly in IPF. GORD can lead to micro-aspirations into the lungs, which may contribute to disease development or progression.
Types of ILD
The most common ILDs include:
- Idiopathic pulmonary fibrosis
- Sarcoidosis
- Extrinsic allergic alveolitis (also known as hypersensitivity pneumonitis)
- Interstitial lung disease associated with connective tissue disease
- Pneumoconiosis
- Interstitial lung disease caused by certain drugs used to treat other conditions
Patient outcomes for ILD
Progressive pulmonary fibrosis is a life-limiting condition with no cure at present. Life expectancy for varies based on several factors, including the type of pulmonary fibrosis, the individual’s overall health, age, and whether they receive treatment.
For idiopathic pulmonary fibrosis (IPF), the average life expectancy is generally cited as 3 to 5 years after diagnosis without treatment. However, this figure is based on older data, and newer treatments like antifibrotic medications have been shown to slow disease progression, potentially extending life expectancy beyond these earlier estimates. Some individuals may live significantly longer with treatment and good management.
Living with pulmonary fibrosis

Watch John's story of living with IPF
